
|
Photophysics and Photochemistry of Transition Metal Compounds |
| Home Research Members Collaborations Publications |



|
||||||||
The ability of different density functionals to describe the structural and energy differences between the high- [5T2g:(t2g)4(eg)2] and low- [1A1g:(t2g)6(eg)0] spin states of small octahedral ferrous compounds is studied. This work is an extension of our previous study of the hexaquoferrous cation, [Fe(H2O)6]2+, [J. Chem. Phys. 120, 9473 (2004)] to include a second compound—namely, the hexaminoferrous cation, [Fe(NH3)6]2+—and several additional functionals. In particular, the present study includes the highly parametrized generalized gradient approximations (GGAs) known as HCTH and the meta-GGA VSXC [which together we refer to as highly parametrized density functionals (HPDFs)], now readily available in the GAUSSIAN03 program, as well as the hybrid functional PBE0. Since there are very few experimental results for these molecules with which to compare, comparison is made with best estimates obtained from second-order perturbation theory-corrected complete active space self-consistent field (CASPT2) calculations, with spectroscopy oriented configuration interaction (SORCI) calculations, and with ligand field theory (LFT) estimations. While CASPT2 and SORCI are among the most reliable ab initio methods available for this type of problem, LFT embodies many decades of empirical experience. These three methods are found to give coherent results and provide best estimates of the adiabatic low-spin–high-spin energy difference, ΔELHadia, of 12 000–13 000 cm−1 for [Fe(H2O)6]2+ and 9 000–11 000 cm−1 for [Fe(NH3)6]2+. All functionals beyond the purely local approximation produce reasonably good geometries, so long as adequate basis sets are used. In contrast, the energy splitting, ΔELHadia, is much more sensitive to the choice of functional. The local density approximation severely over stabilizes the low-spin state with respect to the high-spin state. This “density functional theory (DFT) spin pairing-energy problem” persists, but is reduced, for traditional GGAs. In contrast the hybrid functional B3LYP underestimates ΔELHadia by a few thousands of wave numbers. The RPBE GGA of Hammer, Hansen, and Nørskov gives good results for ΔELHadia as do the HPDFs, especially the VSXC functional. Surprisingly the HCTH functionals actually over correct the DFT spin pairing-energy problem, destabilizing the low-spin state relative to the high-spin state. Best agreement is found for the hybrid functional PBE0. | ||||||||
|
 |
|||||||
In the iron(II) low-spin complex [Fe(bpy)3]2+, the zero-point energy difference between the 5T2g(t42ge2g) high-spin and the 1A1g(t62g) low-spin states, ΔE0HL, is estimated to lie in the range of 2500-5000 cm-1. This estimate is based on the low-temperature dynamics of the high-spin→low-spin relaxation following the light-induced population of the high-spin state and on the assumption that the bond-length difference between the two states ΔrHL is equal to the average value of ≈0.2 Å, as found experimentally for the spin-crossover system. Calculations based on density functional theory (DFT) validate the structural assumption insofar as the low-spin-state optimised geometries are found to be in very good agreement with the experimental X-ray structure of the complex and the predicted high-spin geometries are all very close to one another for a whole series of common GGA (PB86, PW91, PBE, RPBE) and hybrid (B3LYP, B3LYP*, PBE1PBE) functionals. This confirmation of the structural assumption underlying the estimation of ΔE0HL from experimental relaxation rate constants permits us to use this value to assess the ability of the density functionals for the calculation of the energy difference between the HS and LS states. Since the different functionals give values from -1000 to 12000 cm-1, the comparison of the calculated values with the experimental estimate thus provides a stringent criterion for the performance of a given functional. Based on this comparison the RPBE and B3LYP* functionals give the best agreement with experiment. | ||||||||
 |
|
|||||||
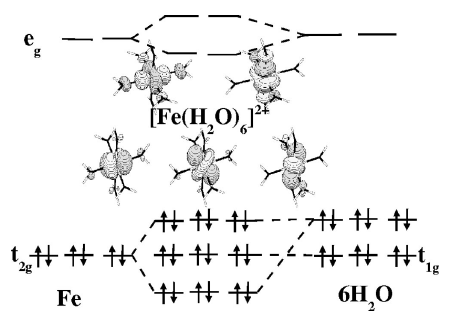
A comparison of density functionals is made for the calculation of energy and geometry differences for the high- [5T2g: (t2g)4(eg)2] and low- [1A1g: (t2g)6(eg)0] spin states of the hexaquoferrous cation [Fe(H2O)6]2+. Since very little experimental results are available (except for crystal structures involving the cation in its high-spin state), the primary comparison is with our own complete active-space self-consistent field (CASSCF), second-order perturbation theory-corrected complete active-space self-consistent field (CASPT2), and spectroscopy-oriented configuration interaction (SORCI) calculations. We find that generalized gradient approximations (GGAs) and the B3LYP hybrid functional provide geometries in good agreement with experiment and with our CASSCF calculations provided sufficiently extended basis sets are used (i.e., polarization functions on the iron and polarization and diffuse functions on the water molecules). In contrast, CASPT2 calculations of the low-spin–high-spin energy difference ΔELH = ELS−EHS appear to be significantly overestimated due to basis set limitations in the sense that the energy difference of the atomic asymptotes (5D→1I excitation of Fe2+) are overestimated by about 3000 cm−1. An empirical shift of the molecular ΔELH based upon atomic calculations provides a best estimate of 12 000–13 000 cm−1. Our unshifted SORCI result is 13 300 cm−1, consistent with previous comparisons between SORCI and experimental excitation energies which suggest that no such empirical shift is needed in conjunction with this method. In contrast, after estimation of incomplete basis set effects, GGAs with one exception underestimate this value by 3000–4000 cm−1 while the B3LYP functional underestimates it by only about 1000 cm−1. The exception is the GGA functional RPBE which appears to perform as well as or better than the B3LYP functional for the properties studied here. In order to obtain a best estimate of the molecular ΔELH within the context of density functional theory (DFT) calculations we have also performed atomic excitation energy calculations using the multiplet sum method. These atomic DFT calculations suggest that no empirical correction is needed for the DFT calculations. | ||||||||
Download this list in format RIS
 EndNote
EndNote  BibTex
BibTex  PDF XML
PDF XML Last update Friday December 08 2017